Hemokromatoz, ince bağırsak tarafından uygunsuz şekilde fazla demir emiliminin gerçekleştiği genetik bir hastalıktır. Fazla demir; karaciğerde, pankreasta, dalakta, tiroid bezinde, deride, kalpte, eklemlerde, yumurtalıklarda, testislerde ve ön hipofiz bezinde birikir. Tedavi edilmeyen kişilerin erken dönemdeki bulguları arasında; karın ağrısı, halsizlik, kilo kaybı, eklem ağrıları yer alabilir. Bazı hastalarda aşırı demir yüküne bağlı olarak organ hasarı gelişir ve bunun sonucunda kardiyomiyopati, artropati, diabetes mellitus, siroz ile karaciğer ve pankreas kanseri gibi risklerde ciddi bir artış görülebilir.
Hemokromatoz Hastalığı tedavi edilmeyen kişilerin erken dönemdeki bulguları arasında; karın ağrısı, halsizlik, kilo kaybı, eklem ağrıları yer alabilir.
Otozomal resesif şekilde kalıtılan hemokromatoz, HFE genindeki patojenik varyantlar sonucunda ortaya çıkmaktadır. Bu patojenik varyantlar HFE geni tarafından kodlanan HFE proteininin işlevinin azalmasına ya da tamamen kaybolmasına yol açar. Hemokromatoz ile ilişkili patojenik varyantlar arasında en sık rastlanılanlar C282Y ve H63D varyantlarıdır. Bunlar dışında S65C ve E168X varyantları da daha ender görülmekle birlikte hemokromatoz hastalığının gelişmesi ile bağlantılı bulunmuşlardır. Hastaların %90 – 100 kadarı patojenik varyantları homozigot durumda (çift kopya) taşır, fakat tek kopyaya sahip kişilerin bile demir metabolizmalarında ufak bozukluklar ortaya çıkabilmektedir.
Hemokromatoz Hastalığı Moleküler Genetik Analizi
Hemokromatoz ile ilişkili HFE genindeki bu patojenik varyantlar, moleküler genetik analiz ile saptanabilir. Hastalık, klinik olarak ortaya çıkan bulgular ile teşhis edildiğinde genellikle vücutta meydana gelen hasar artmış olur ve böyle bir durumda tedavi yalnızca kısmen başarılı olabilmektedir. Ancak moleküler genetik analiz ile erken tanı koyulabildiğinde, tedaviye erken başlanarak hastalığa bağlı sorunları azaltmak ve / veya geciktirmek mümkün olabilir.
Meme/over kanseri dünyada en sık rastlanan kanser türleri arasında yer almaktadır. Toplum genelinde, kadınların yaklaşık %12 kadarının meme kanserine yakalanacağı öngörülmektedir. Meme ve/veya over kanserine kalıtsal yatkınlık ile ilgili olduğu bulunan pek çok gen tespit edilmiştir. Bu genler arasında yer alan BRCA1 ve BRCA2 genlerindeki mutasyonların, meme/over kanseri geliştirme riskini %50-85 kadar arttırdığı bildirilmiştir.
MEME KANSERİ
Kadınlarda teşhis edilen kanserlerin %25’ini meme kanseri vakaları oluşturur. Tüm meme kanserlerinin %5-10’unun kalıtımsal olduğu ve BRCA1 ve BRCA2 genlerindeki mutasyonların da meme kanseri riskini en çok arttıran mutasyonlar olduğu gösterilmiştir. BRCA genlerindeki mutasyonlar, sadece hastanın genetik yatkınlığının göstergesidir. Yaşam tarzının ve çevresel etkenlerin de önemli bir rolü olduğu unutulmamalıdır. Dolayısıyla, BRCA1 ve BRCA2 mutasyonu tespit edilmiş bir kişide, meme veya over kanserinin ya da başka bir kanser türünün mutlaka ortaya çıkacağını ifade etmek hatalı bir yaklaşım olacaktır. Buna karşılık, mutasyon taşımayan bir kişide sporadik olarak meme kanseri gelişebilir.
BRCA1 ve BRCA2 GENLERİNİN ROLÜ
DNA hasarını tamir etmekle görevli olan BRCA1 ve BRCA2 genleri, tümör baskılayıcı genlerdir. Mutasyona uğradıklarında işlevlerini kaybederler ve doğal fonksiyonlarının tam aksi yönünde kanser oluşumuna sebep olan bir faktör haline gelirler. BRCA1 ve BRCA2 genlerinde, toplam 2600’den fazla mutasyon tanımlanmıştır. Bu mutasyonlar otozomal dominant şekilde kalıtılırlar ve yüksek derecede penetrans gösterirler. BRCA1 ve BRCA2 mutasyonu taşıyıcılarının toplumlardaki genel sıklığı yaklaşık 1/500 – 1/1000’dir.
BRCA1 veya BRCA2 geninde kalıtımsal bir mutasyon taşımak, meme, over ve bazı diğer kanserlerin oluşma riskini, hem kadınlarda hem de erkeklerde önemli derecede arttırmaktadır. (Tablo 1)
HANGİ VAKALAR İÇİN GENETİK TEST DÜŞÜNÜLMELİDİR?
Eğer hastanın kendisinin veya ailesinin hikayesi aşağıdaki bulgulardan herhangi birini içeriyor ise genetik test düşünülür:
•Aynı bireyde veya ailenin aynı tarafında pankreas kanseri ile birlikte meme veya over kanseri
•Birisi 50 yaşından genç olmak üzere 2 veya daha fazla akrabada meme kanseri
•Herhangi yaştaki 3 veya daha fazla akrabada meme kanseri
•Ailede daha önceden tanımlanmış mutasyon
Detaylı ve hatasız bir aile hikayesinin çıkarılması, kişinin kalıtımsal meme veya over kanseri riskinin belirlenmesinde çok büyük önem taşır. Kanseri erken teşhis edebilmek için atılacak her adımla, hastanın önleyici tedbirlere ve proaktif tedavilere ulaşması sağlanmış olacaktır. Sonuç olarak hastalığın daha iyi prognoz göstermesini sağlamak mümkün olacaktır.
(Risk kriterlerini açıklayan daha detaylı bilgiye “NCCN Clinical Practice Guidelines For Breast Cancer” ve benzeri kaynaklardan ulaşılabilir.)
1. Kalıtımsal meme/over kanseri için orta derece risk taşıyan vaka örneği:
Ailenin aynı tarafında, 70 yaşından erken kansere yakalanmış, iki tane 1. ve 2. dereceden akraba bulunmaktadır.
2. Kalıtımsal meme/over kanseri için yüksek derece risk taşıyan vaka örneği:
Ailede 1. ve 2. Dereceden bilateral meme kanseri olan (biri 50 yaşından önce) ve over kanseri olan akrabalar bulunmaktadır.
BRCA1 ve BRCA2 GENETİK ANALİZLERİ
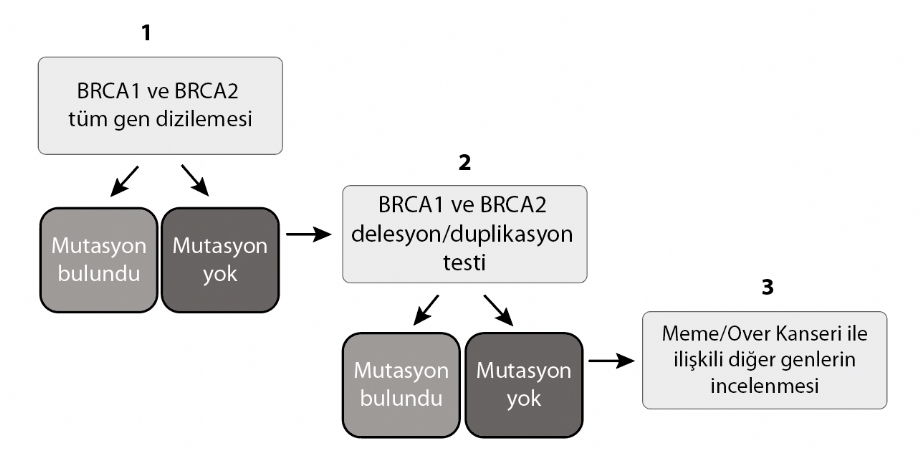
1) BRCA1 ve BRCA2 Tüm Gen Dizilemesi
Aile hikayesinin ve/veya klinik tablonun kalıtımsal meme/over kanseri riskini işaret ettiği vakalarda, BRCA1 ve BRCA2 genlerinde Tüm Gen Dizilemesi yapılmaktadır. Yapılan dizi analizi sonucunda bir mutasyon tespit edilmesi halinde, hastanın ailesindeki diğer bireylere de bu ailesel mutasyon için test yapılması önerilir.
2) BRCA1 ve BRCA2 Delesyon/Duplikasyon Testi
Bazı durumlarda mutasyonlar, az sayıdaki DNA bazlarında değil, genin bir bölgesinin tamamının delesyonu veya duplikasyonu şeklinde ortaya çıkabilir. Bu tip büyük delesyon/duplikasyon mutasyonlarının BRCA1 ve BRCA2 genlerinde oldukça sıklıkla meydana geldiği görülmüştür. Ancak bu tür mutasyonlar, Tüm Gen Dizilemesi ile net olarak tespit edilemez. Bu mutasyonların analizi için MLPA yöntemi ile Delesyon/Duplikasyon Testi yapılması gerekir.
3) Meme/Over Kanseri ile İlişkili Diğer Genlerin İncelenmesi
BRCA 1 ve BRCA 2 dışında meme ve/veya over kanseri geliştirme riski ile bağlantılı oldukları saptanan başka genlerinde sayısı giderek artmaktadır. Diğer genlerdeki kalıtımsal mutasyonlar sadece meme ve /veya over kanserlerinin değil, aynı zamanda melanoma, kolon, pankreas ve prostat gibi başka kanserlerin riskinide arttırmaktadır. BRCA1 ve BRCA2 genlerinde yapılan testlerde mutasyon bulunmayan, fakat meme ve/veya over kanseri açısından kuvvetli bir aile hikayesine sahip olan kadınlar için Yeni Nesil Dizileme (Next Generation Sequencing – NGS) yöntemiyle yapılan paneller önerilebilir. Bu panellerde, meme ve/veya over kanseri ile ilgisi bulunmuş diğer yatkınlık genleri taranmaktadır. NGS yöntemi ile yapılan bu geniş kapsamlı dizilemeler sonucunda eğer mutasyon tespit edilirse, elde edilen bulguların Sanger Dizileme Yöntemi ile doğrulanması tavsiye edilir.
https://birunigenetik.com.tr/wp-content/uploads/2021/11/brca1-brca2-genetik-testleri.jpg400600İskender Maraşhttps://birunigenetik.com.tr/wp-content/uploads/2022/05/Biruni-Genetik-Logo-yeni.pngİskender Maraş2024-06-27 16:16:132024-09-25 14:09:32BRCA1 ve BRCA2 Genetik Testleri
İlaç seçimi, ilaç dozu, etkileşimi ve yan etki değerlendirmesinde, kişiye özel tedavi kararınızda genetik desteğiniz.
İlaçların istenilen etkinliği göstermemesi ve ilaca bağlı yan etkilerin en önemli nedeni, kullanılan ilaçların hastanın genetik yapısına uygun olmamasıdır.
İlaçların metabolizması kişiden kişiye farklılık göstermektedir. Bu durum ilaçların metabolizma yolağındaki enzimlerin, her kişide farklı genetik yapılarla düzenlenmiş olmasına bağlıdır. Aynı etken madde farklı kişilerde yavaş, hızlı veya normal metabolize edilmesine göre az, çok veya normal etkinlik göstererek her bireyde farklı tedavi sonuçlarına ve yan etkilere yol açar.
Uygulanan tüm ilaç tedavilerinde, vakaların % 40-70’inde ilaçlar yeteri kadar etkili olamamakta ve hastaların %7-10’unda ilaçların yan etkileriyle karşılaşılmaktadır. Her yıl ABD’de ilaçlara bağlı en az 2,2 milyon yan etki vakası oluşmakta ve bunların 100.000’den fazlası ölümle sonuçlanmaktadır. Bir ilacın hangi hastada etki veya yan etki oluşturacağını gösteren Farmakogenetik testlerin kullanımı yan etki vakalarını ve bunlara bağlı kayıpları azaltmaktadır.
Farmakogenetik testler bir hastanın ilaç tedavisine vereceği yanıt, toksisite ve ilaç etkileşimlerini belirlemek için yapılan DNA analizleridir. Kişiye özel tedavi uygulamasına olanak sağlar.
Amerika Birleşik Devletleri’nde sağlık otoritesi FDA ve Avrupa’da EMA en sık kullanılan yüzlerce ilacın prospektüsünde Farmakogenetik bilgi ve özel uyarılar bulunmasını zorunlu kılmaktadır. Bu ilaçlarla tedaviye başlanırken kişinin genotipine özel doz ayarlaması, klinik yanıt varyasyonları ve yan etki risklerinin belirlenebilmesi için Farmakogenetik test yapılması istenmektedir.
FARMAKOGENETİK DNA PANELİ:
• Uygun etken madde seçimi,
• Optimal dozun belirlenmesi,
• İlaç etkileşimlerinin belirlenebilmesi,
• Yan etki olasılıklarının azaltılması
• Tedaviye uyumun artırılması
sağlanarak kişiye özel tedavi planlaması ve uygulaması mümkün olur.
Test prosedürü kolay ve hızlıdır.
Ağız içinden epitel örneği alınır. İğne ve kan örneği yoktur.
Alınan örnek özel kitine yerleştirilir.
Güvenlik barkodları ile korumaya alınır ve uygun şartlarda laboratuvara iletilir.
Genetik konsültasyon içeren sonuç kısa bir sürede gönderilir.
Bilinen tüm klinik fenotiplerle ilişkili genler hedefimizde
Klinik Ekzom testi, bilinen klinik fenotiplerle ilişkilendirilmiş bütün genlerin kodlayıcı bölgelerine odaklanır. Böylelikle ek maliyetleri azaltır ve genlerin hastalıklarla ilişkili olmayan bölgelerinin dizilenmesiyle ortaya çıkabilecek belirsiz sonuçlara engel olur.
Mevcut en geniş kapsamlı NGS (Yeni Nesil Dizileme) Paneli olan Klinik Ekzom testinin içeriğinde 3200’den fazla hastalık bulunmaktadır. OMIM ve HGMD’de net olarak genotip–fenotip ilişkisi ortaya konmuş hastalıkları hedefler.
Klinik Ekzom içeriğindeki yaklaşık 6700 gen, özel tasarlanmış problar sayesinde güçlü bir performans ile kısa sürede dizilenmektedir. Dizilenenler arasındaki yaklaşık 4000 genin kodlayıcı bölgeleri %100 oranında kapsanmaktadır.
Geniş kapsamlı klinik bilgi imkanı
Yoğun içerik
Klinik fenotiplerle ilgili bilinen tüm genler hedeflenir
Herhangi bir kırpılma hatasını saptayabilmek üzere +/- 10bp intron bölgeleri değerlendirilir
En iyi panel özellikleri
Hedeflenen bazların %95’i > 20X kapsanır
Yaklaşık bir ayda sonuçlandırılır
Raporlanan tüm mutasyonlar Sanger dizileme ile doğrulanır
Yüksek yetkinlik
En ileri biyoinformatik süreçler
Klinik verinin ayrıntılı değerlendirmesi
Açık klinik tanı raporu
Negatif vakalarda ayırıcı tanıya dair tavsiyeler
Az örnek gereksinimi
1 ml EDTA tam kan
1µg saflaştırılmış DNA
Klinik ekzom testi kapsamındaki hastalıklar
Klinik ekzom testinden kimler faydalanabilir?
Klinikle ilişkili genleri daha derinden incelemeyi isteyen hekimler
Tanımlanamamış fenotipi olan hastalar
Fenotipik heterojeniteyle karşı karşıya olan ve tüm ekzom testi için imkanı olmayan hekimler
Tanımlanmış fenotipi olup hastalığa özgü pahalı NGS paneller yerine uygun bir seçenek arayan hastalar
Farklı testlerden elde edilecek verilerin karşılaştırması
Trombofili, kanda pıhtılaşma eğiliminin arttığı, dolayısı ile Venöz Tromboemboli (VTE) riskinin yüksek olduğu durumları tanımlamak için kullanılan bir terimdir. Günümüzde VTE Batı ülkelerinin genel nüfusunda her yıl yaklaşık 1000’de 1-2 bireyi etkileyen ciddi bir sağlık problemidir (1,2). Çok sayıda edinsel (Tablo 1) ve kalıtsal faktörün (Tablo 2) değişik mekanizmalarla tromboz oluşumuna neden olduğu bilinmektedir (3,4). Arteriyel sistemde endotel hasarının ve trombositlerin fonksiyonel bozukluklarının önemli rol oynadığı, venöz sistemde ise daha çok staz ve pıhtılaşma sistemine ait bozuklukların tromboz gelişimine neden olduğu bilinmektedir (5).
Kalıtsal trombofili nedenlerini genetik olarak taşıyan bireylerde tromboz riski artmakla birlikte, yaşamları boyunca hiçbir trombotik atak geçirmemeleri de mümkündür. Bu kişilerde tekrarlayan trombotik ataklar arasında uzun süren asemptomatik dönemler de olabilmektedir. Bu durum, tek başına kalıtsal nedenlerin yeterli olmadığını, tromboz gelişiminde bazı edinsel faktörlerin katkısı olduğunu göstermektedir. (6,7).
Tablo 1. Edinsel trombofili nedenleri
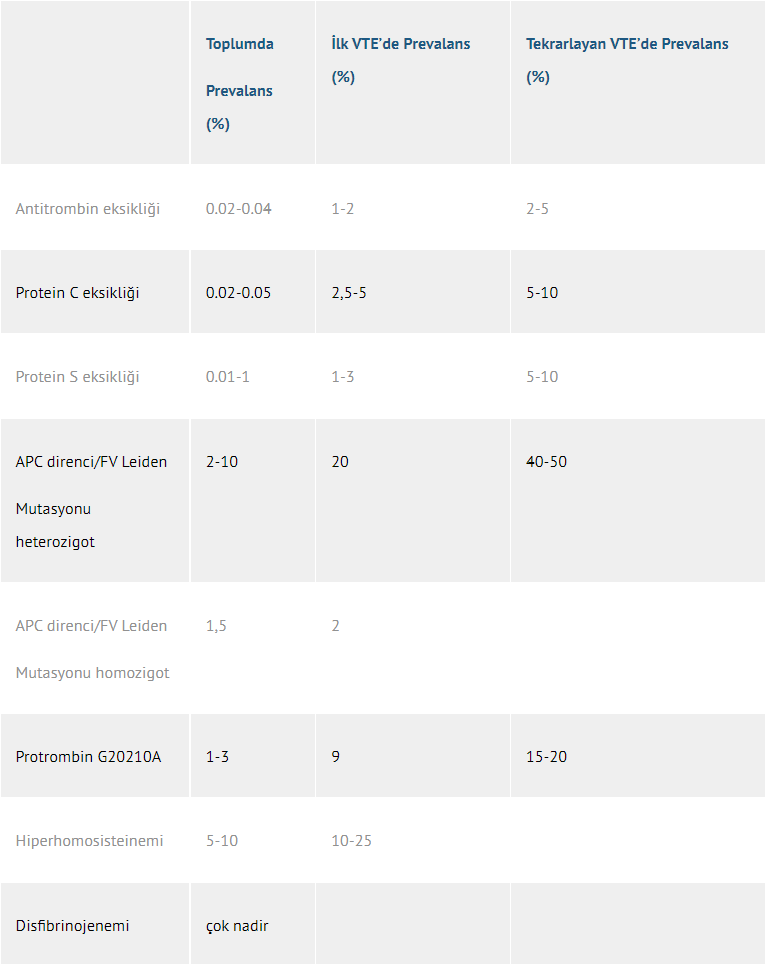
Tablo 2. Kalıtsal trombofili nedenleri ve sıklıkları
Son yirmi yıl boyunca trombofili etyolojisi hakkındaki bilgilerimiz önemli ölçüde artmış ve çeşitli kalıtsal ve edinsel risk faktörleri keşfedilmiştir. Bu durum trombofilinin geniş çapta laboratuvar incelemesine olanak tanımıştır. Trombofilinin laboratuvar incelemesi, APCR, Faktör V Leiden mutasyonu, Protrombin G20210A mutasyonu, fizyolojik antikoagülanlar olan Antitrombin, Protein C ve Protein S eksiklikleri, Antifosfolipid antikorlarının varlığı ve artmış plazma Homosistein düzeylerinin araştırılması suretiyle venöz tromboembolizmin kalıtsal veya edinsel sebeplerini tesbit etmeyi amaçlar(3). Faktör V Leiden mutasyonunun coğrafi dağılımı değişik olup, ülkemizde görülme oranı %7-10’dur.
Kalıtsal trombofili taranması önerilen özellikli hasta grubu:
1- İlk VTE atağını 40 yaş altında geçiren kişiler (8,9,10)
2- Ailesinde tromboembolizm öyküsü olan kişiler
3- Tekrarlayan VTE öyküsü olan hastalar
4- Portal, mezenterik, splenik, hepatik, renal veya serebral venler gibi beklenmedik bölgelerde
tromboz meydana gelmiş kişiler (9,11)
5- Purpura fulminans ile başvuran çocuklar
6- Venöz tromboz riski taşıyan gebeler
7- Warfarine bağlı deri nekrozu öyküsü olanlar
8- Neonatal tromboz öyküsü olan hastalar (3,4)
Protein C ve Protein S testleri trombozun akut döneminde yapıldığında yanlışlıkla düşük bulunabilir. Heparin tedavisi altındaki hastalarda da antitrombin düşük bulunacaktır.
Faktör V Leiden, Protrombin G20210A mutasyonu moleküler inceleme testleridir. APC direnci moleküler bir inceleme olmayıp çabuk sonuçlanan koagülasyon testine dayalı bir incelemedir. APC direncinin <0,86 olduğu hastalarda öncelikle Faktör V Leiden mutasyonu düşünülmelidir.
Yapılan tahlilleri klinikle ilişkilendirebilmek ve sonuçların yanlış yorumlanmasını önleyebilmek için trombofilinin laboratuvar incelemesinde tahlil yapılacak hasta, yapılacak tahliller, tahlil zamanı ve tahlil yöntemi özenle seçilmelidir.
2. Bronic A. Thromboembolic diseases as biological and clinical syndrome – role of the Mediterranean League against Thromboembolic Diseases. Biochem Med 2010; 20: 9–12.
3. Margetic S. Laboratory investigation of trombophilia. J Med Biochem 33: 28-46, 2014.
4. Türk Hematoloji Derneği Kalıtsal Trombofili Tanı ve Tedavi Kılavuzu.
6. Lane DA, Manucci PM, Bauer KA.: Inherited thrombophilia: Part-1. Thromb Haemost 76: 651, 1996.
7. Makris M, Rosendaal FR, Preston FE: Familial thrombophilia: Genetic risk factors and management. J Int Med 242: 9, 1997.
8. Carraro P. European Communities Confederation of Clinical Chemistry and Laboratory Medicine (EC4) Working Group on Guidelines for Investigation of Disease. Guidelines for the laboratory investigation of inherited thrombophilias. Recommendations for the first level clinical laboratories. Clin Chem Lab Med 2003; 41: 382–91.
9. Nicolaides AN, Breddin HK, Carpenter P, Coccheri S, Conard J, De Stefano V, et al. European Genetics Foundation Cardiovascular Disease Educational and Research Trust International Union of Angiology Mediterranean League on Thromboembolism. Thrombophilia and venous thromboembolism. International consensus statement. Guidelines according to scientific evidence. Int Angiol 2005; 24: 1–26.
10. Baglin T, Gray E, Greaves M, Hunt BJ, Keeling D, Machin S, et al. Clinical guidelines for testing for heritable thrombophilia. Br J Haematol 2010; 149: 209–20.
11. College of American Pathologists Consensus Conference XXXVI: Diagnostic issues in thrombophilia. Arch Pathol Lab Med 2002; 126: 1277–433.
MODY (Maturity-Onset Diabetes of Young); genç başlayan (genellikle < 25 yaş), insüline bağımlı olmayan ve klinik olarak heterojen bir diyabet tipidir. Monogenik bir hastalık olan MODY, çoğunlukla otozomal dominant kalıtım gösterir ve tüm diyabet olgularının yaklaşık %2-3’ünü oluşturur. Genetik heterojenite de gösteren MODY’i Tip 1 ve 2 Diabetes Mellitus’tan (DM) ayırt etmek zor olabilmektedir. Birçok toplumda Tip 2 DM tanısı alan hastaların %5’lik bir kısmının MODY olduğu öngörülmektedir. Örneğin İngiltere’de MODY vakalarının %80’den fazlasının Tip 1 veya 2 DM olarak yanlış teşhis edildiği gösterilmiştir. MODY’nin DM’den farklı olan belirgin özellikleri; Tip 1 DM’de sıklıkla tespit edilen otoantikorların ve Tip 2 DM’de genellikle görülen obezitenin bulunmamasıdır. MODY’nin tedavisinin genetik etiyolojiye göre değişiyor olması, seyrek görülen bu hastalığın klinik önemini arttırmaktadır.
MODY Temel Panel hastalık ile ilişkilendirilmiş HNF1A, HNF1B, HNF4A ve GCK genlerinin dizilenmesini içerir.
MODY Genişletilmiş Panel kapsamında ise temel paneldeki genlere ek olarak ABCC8, INS, KCNJ11, PDX1, NEUROD1, KLF11, CEL, PAX4 ve BLK genleri yer almaktadır.
MODY panellerinde, belirtilen genlere ait tüm kodlayan ekzonik bölgeler yeni nesil dizileme ile taranır ve ortaya çıkan varyasyonların biyoinformatik analizleri yapılır. Ortalama hizmet süresi 4-6 haftadır.
Gençlerde görülen ve erişkin başlangıçlı diyabet gibi seyreden MODY şüphesi olan hastaların genellikle:
Yaşları genç olur (diyabet başlangıç yaşı <25)
Ailelerinde iki veya daha fazla kuşakta diyabet bulunur (otozomal dominant geçişli)
Hiperglisemiye eşlik eden obezite veya metabolik sendrom öyküsü bulunmaz
İnsülin dirençleri yoktur ve pankreas rezervleri iyidir
Asıl sorun insülin sekresyon mekanizmasında olduğu için otoantikorları negatif bulunur
Kan glukoz regülasyonları için insülin tedavisi gerekmez veya düşük dozla regülasyon sağlanır
Günümüzde birçok vakada MODY tanısı atlanmış veya Tip 1 veya 2 DM olarak yanlış sınıflandırılmıştır. Tip 1 ve 2 DM tanısı ile izlenen, ancak atipik gidiş gösteren, insülin direnci saptanmayan veya sülfonilüre grubu ilaçlara aşırı duyarlılığı olan hastalarda MODY’yi düşünmek ve uygun moleküler testler ile tanıyı doğrulamak, diyabet tedavisi için anahtar rol oynamaktadır. Moleküler tanı, hastanın prognozunu etkileyecek uygun tedavinin seçimi, genetik danışma verilmesi ve risk altındaki bireylerin taranması açısından çok büyük önem taşımaktadır. Hastalığın seyri ve ek komplikasyonlar ile ilişkili risk, genellikle ilişkili gen mutasyonlarına bağlıdır. Gelişen moleküler test olanakları ile yeni genler tanımlandıkça, MODY sınıfının giderek genişleyeceği düşünülmektedir. MODY tanısı için, genetik test genellikle sadece klasik özelliklere sahip olanlarda yapılmaktadır. Bununla birlikte, genetik olarak MODY tanısı konan hastaların sadece %50’si klasik kriterleri karşılamaktadır.
MODY (OMIM: 606391) hastalarının %80-85’lik bir kısmında HNF1A, HNF1B, HNF4A ve GCK genlerinden birinde mutasyonlar görülmektedir. Diğer genlerdeki mutasyonlar (NEUROD1, KLF11, CEL, PAX4, PDX1, INS, BLK, KCNJ11 ve APPL1) ise MODYhastalarının %15-20’lik kısmında görülmektedir. Ek olarak, ABCC8 genindeki mutasyonların da MODY kriterlerini karşıladığı gösterilmiştir.
HNF1A ve HNF4A mutasyonları olan hastalar, yavaş ilerleyen beta-hücre işlev bozukluğuna sahiptirler. Düşük doz sülfonilüre ile tedavi, insülin veya diğer tedavilere kıyasla stabil glisemik kontrol ve yaşam kalitesinde iyileşme ile sonuçlanmaktadır. HNF1Agenindeki mutasyonlar, MODY olgularının yaklaşık %20-50’sini oluşturan MODY3’e neden olur. MODY3 ile ilişkili fenotip, hastalarda normal kan glikoz seviyelerinden yüksek seviyelere kadar değişkenlik göstermektedir. MODY3 hastaları, erken yaşta beta hücre yetmezliği ve ilerleyici hiperglisemi veya ergenlik döneminde mikrovasküler ve makrovasküler komplikasyon riski altındadırlar. Hiperglisemik kontrol sülfonilüre tedavisi ile uzun yıllar sürdürülebilir, ancak MODY3’lü bireyler genellikle insülin tedavisine ihtiyaç duyabilirler. HNF4A genindeki mutasyonlar, MODY olgularının yaklaşık %5’ini oluşturan MODY1’e neden olur ve MODY3’e benzer bir seyir gösterir.
PDX1 genindeki heterozigot mutasyonlar ise MODY4’e neden olur. MODY2, MODY olgularının %20-50’sini oluşturan GCK genindeki mutasyonlar sonucu ortaya çıkar. MODY2 hastaları tipik olarak, hafif-orta derecede açlık hiperglisemisine sahiptirler ve genellikle asemptomatiktirler. MODY2’ye sahip bireylerin çoğunluğu tek başına diyetle kontrol altına alınır ve nadiren ilaç tedavisine ihtiyaç duyulmaktadır. GCK-MODY, tipik olarak HbA1c <%7 (53 mmol/mol) ile birlikte hafif ve ilerleyici olmayan hiperglisemi gösteren bir fenotipe sahiptir. Diğer tiplerdeki artmış mikrovasküler ve makrovasküler komplikasyonlar ile ilişkili değildir. Tedavi genellikle HbA1c düzeyini değiştirmez. GCK-MODY’nin moleküler tanısı, farmakolojik tedavinin kesilmesine ve gerekli tıbbi gözetim sıklığının azalmasına olanak tanır.